基本原理
对于取代基类型不同但其他结构相同的反应物,反应的活化自由能与吉布斯自由能地差成一定的比例关系。路易斯·普拉克·哈米特于1935年提出此概念,并于1937年首先提出方程的形式。
在芳香族化合物,尤其是取代苯的反应中,k0是不含取代基反应物的反应速率常数,k是含有取代基的反应物的速率常数。它们之间存在如下关系:σ称为“取代基常数”,只与取代基的类型有关;ρ称为“反应常数”,只与反应的类型有关,与取代基类型无关。哈米特方程的另一种形式是描述反应中平衡常数的关系,即:
K0是不含取代基反应物的反应平衡常数,K是含有取代基的反应物的反应平衡常数。
哈米特方程结构图
表达反应速率与反应物结构间定量关系的一个方程式。1937年由L.P.哈米特首先提出。该方程在有机结构理论中很重要。在芳香族化合物,特别是苯类化合物的反应中,若不含取代基的反应物的反应速率常数为k0,当引入取代基后,其反应速率常数为k,则k与k0之间有如下关系:
lgk=lgk0+σρ
这就是哈米特方程。式中σ为取代常数,其值决定于取代基团的性质和位置(如邻位、间位、对位);ρ为常数,决定于反应类型。
哈米特方程的另一种形式是描述反应平衡常数的关系,即:
lgK=lgK0+σρ
式中K0为不含取代基的反应物的平衡常数;K为含取代基的反应物的平衡常数。
规定氢的σ值为0.00,苯甲酸和取代苯甲酸的水溶液在25℃时电离反应的ρ为1。根据苯甲酸的电离常数K和取代苯甲酸的电离常数K,即可求得各种取代基的相对σ值:
表1列出了一部分基团的σ值。给电子基团的σ为负值,吸电子基团的σ为正值。由σ数值的大小,可定量表示取代基给电子性或吸电子性的强弱。σ是各种效应的综合结果。
根据已求得的σ值,可用哈米特方程求得各反应的ρ值。表2列出了部分反应的ρ值。ρ值可为<0、0、0<ρ<1、≥1等各种可能的数值。人们认为ρ的数值与反应过渡态的荷电情况有关。当过渡态与反应物相比具有正电性时,则ρ0;电性无变化时,则ρ=0。如果与ρ=1的标准反应相比,则ρ1时,则过渡态的电荷改变比标准反应具有更强的负电性,ρ值越大,则负电性越强。测定反应的ρ值,已成为研究有机反应机理的有效手段之一。
取代基常数

哈米特方程分子式
各种取代基常数是通过测量间位、对位取代苯甲酸在25°C水溶液中的pKa值得到的。取代基仅限于羧基的间、对位,因为这些位置对羧基的影响可以假定纯粹是电子效应,而邻位取代则是电子效应和立体效应的综合。脂肪族取代羧酸由于单键可以自由旋转,可以采取的构象众多,存在一定的立体效应,从而降低了过渡态结构和反应平衡位置之间的相关性。氢为取代基时的取代基常数和反应常数被定为1(即苯甲酸自身),其他取代基的取代基常数是在此基础上,通过以下公式计算获得的:
公式表格
| Hammett取代基常数 | | |
| 取代基 | 对位常数 | 间位常数 |
| –NH2 | -0.66 | -0.161 |
| –OCH3 | -0.268 | +0.115 |
| –OC2H5 | -0.25 | +0.15 |
| –N(CH3)2 | -0.205 | -0.211 |
展开表格右表中列出了一些比较常见的取代基常数值。可以看出,取代基常数值是取代基电子效应的量度,其效果是诱导效应(±I)和共轭效应(±M)的综合效果。σ值最大的取代基是氰(qing2)基和硝基,是典型的吸电子基团,相应的芳香羧酸由于阴离子更加稳定,使羧酸酸性更强,pKa更小。
接下来是各种卤素取代基。它们有吸电子的诱导效应和供电子的共轭效应,但总体上仍是吸电子的,故σ仍为正值。对位取代的卤素由于可以和苯环共轭(e4),产生下面的共振式结构,卤素的电子通过苯环向羧基转移,使得质子不易离去,酸性较低。间位取代的卤素由于不存在这个现象,因此σ值大于对位的σ值。
继续往下得到甲氧基和乙氧基等供电子基团,它们共轭效应和诱导效应的方向相反,但诱导效应占主要地位,故σ为负值。烷基和芳基的σ值也为负值,但这些基团两种效应的方向是一致的,另外也存在给电子的超共轭效应。
ρ值
在取得各种取代基常数值后,哈米特又用不同取代的芳香羧酸发生某一特定反应,并计算出这些反应的速率常数。他发现,如果用log(kX/kH)对σ作图(
Hammett图
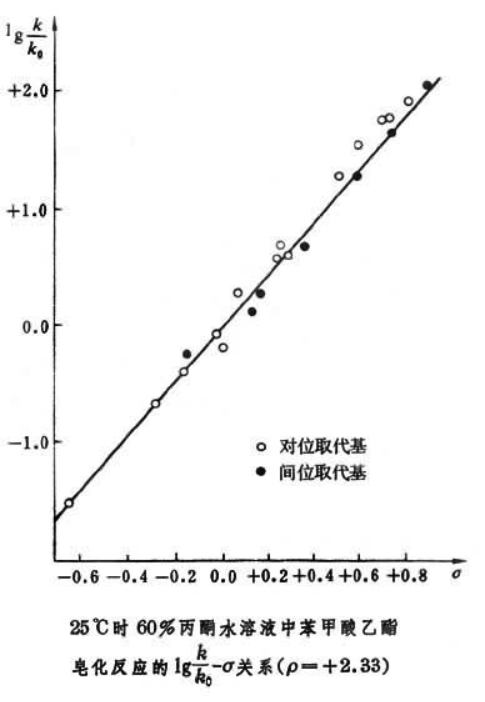
),可以发现两者符合很好的线性关系。log(kX/kH)-σ图的斜率记作ρ,也就是此类型反应的反应常数。苯甲酸解离的反应常数定为1。大部分反应都存在这种线性关系,其中一个比较典型的反应是30°C时,取代苯甲酸乙酯在水/乙醇混合物中发生水解的反应。这个反应的反应常数值为+2.498。
反应常数值
其他很多反应的ρ值都是已知的。以下是哈米特在原始文献中列出的一些反应常数值:
取代肉桂酸酯在乙醇/水混合物中的水解反应(+1.267);取代苯酚在水中的解离反应(+2.008);酸催化下取代苯甲酸在乙醇中的酯化反应(-0.085);酸催化下取代苯乙酮在乙醇/水/盐酸混合物中的溴化反应(+0.417);69.8°C时,取代氯化苄在丙酮/水混合物中的水解反应(-1.875)。由于ρ是由速率商的对数对取代基常数作图得到的,因此一个反应的ρ值可以看作是,相对于苯甲酸解离反应而言,这个反应的反应速率受取代基变化的灵敏度的定量测量。如果吸电子基团对反应起加速作用,很明显ρ>0,哈米特图的斜率是正值;如果给电子基团对反应起加速作用,ρ<0,哈米特图向后倾斜。也就是说:
ρ>1,反应速率比苯甲酸解离的反应更容易受取代基影响,且速率控制步骤是负电荷累积的过程;0<ρ<1,反应速率比苯甲酸解离的反应更不容易受取代基影响,且速率控制步骤是负电荷累积的过程;ρ=0,反应速率不受取代基影响,且速率控制步骤中反应中心电荷无明显变化;ρ<0,速率控制步骤是反应中心正电荷累积的过程。这是哈米特方程用于测定有机反应机理的基础。当一个有机反应存在两个未经证实的机理时,可以通过用不同取代的芳香族化合物原料分别发生反应,计算出反应速率和ρ值,并判断出速率控制步骤时反应中心是否有电荷的明显变化。如果ρ在-1与1之间,则基本上可以否定数控不涉及正/负电荷累积的机理。有趣的是,协同反应(如DA反应)由于基本不涉及电荷变化,故其ρ值大多在-1与1之间,且ρ值的正负可用于判断哪个反应物分子出HOMO,哪一个出LUMO。
特例
有时哈米特图是非线性的,例如它可以是两条线段组成的折线图。这通常预示着反应机理或过渡态位置的改变。比如芳香酰氯的水解反应,它的哈米特图是一个由ρ=-4.4与ρ=+2.5组成的折线图,存在一个最低点。这是因为供电子基团取代的酰氯通过酰基正离子机理反应的比例更大,即首先氯离子离去,然后水进攻生成的正离子;而吸电子基团取代的酰氯则通过亲核酰基取代机理反应,过渡态中负电荷累积,所以ρ值是正的。