
精选百科
本文由作者推荐
色散力
1930年发现的吸引力
发现历史
色散力首次被提出是在1930年,由德国物理学家菲列兹·伦敦(Fritz London,1900-1954)的一篇于1930年论文中提出,因此,色散力又被称为伦敦力。色散力不分散任何东西,而是根据其量子力学处理与光学色散的数学处理的相似性而命名。
定义
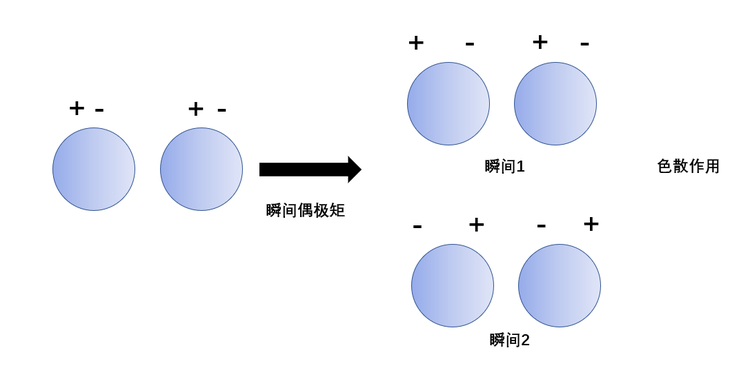
在分子内部,电子和原子核在时刻震动着,过程中它们会发生瞬时的相对位移,瞬间分子的正、负电中心不重合,产生瞬时的偶极矩,进而产生的力叫做色散力。色散力存在于一切分子之间。
原理
分子间的色散力
分子间作用力,或者更广泛地说,分子内部或分子之间的非共价相互作用,统称为范德华瓦尔斯力,其中包含色散力。任何分子都有正、负电中心,非极性分子也有正负电中心。分子的极性和变形性是当分子互相靠近时分子间产生吸引作用的根本原因。两个非极性分子相遇时,分子中的电子和原子核都在不停运动,过程中它们会发生瞬时的相对位移,瞬间分子的正、负电中心不重合。虽然在一段时间内大体上看分子的正、负电中心是重合的,表现为非极性分子,但每一瞬间却总是出现正、负电中心不重合的状态,形成了瞬时偶极。当两个非极性分子靠得较近,如相距非常近时,这两个分子的电中心步调一致地处于异极相邻的状态。这样就在两个分子之间产生一种吸引作用力,叫做色散力。虽然每一瞬间的时间极短,但在下一瞬间仍然重复着这样异极相邻的状态。对于极性分子,其本身就具有不重合的正、负电中心,当非极性分子与它靠近到几百皮米时,在极性分子的电场影响(诱导)下,非极性分子中原来重合着的正、负电中心被极化,产生色散力。而极性与非极性分子相遇时,每种分子都有变形性,自然会有色散力。色散力是电磁作用的主要结果,但却不能用经典力学描述,完全是量子力学效应 。 因此对色散能的严格计算比其它两种分子间能要更复杂。
原子间的色散力
对于原子A与原子B的相互作用,典型的论证大致如下:A的瞬时波动产生偶极矩,这个偶极矩在B中诱导了一个有利的偶极矩,这两个瞬时偶极子之间的相互作用是吸引的。B的瞬时波动也会在A中诱导有利的偶极子,这些也有助于形成吸引人的相互作用势。色散因此被描述为相互诱导偶极矩的相互作用。这个论点没有错,但它不是电子基态色散力的正确描述。事实上,这些力在基态下不随时间变化或波动;色散力完全可以用与时间无关的量子力学来理解,量子力学具有不随时间波动的定态,而上述描述它们的方式可能会使人认为色散力确实与时间有关。
特点
特征
色散力,就像势能的特征一样,可以用与时间无关的量子力学来描述,它们是与电子相关和量子力学叠加的表现。但并不意味着色散力的产生需要随时间变化的电子密度分布。色散作用与分子的极化率有关,极化率愈大的分子之间色散作用愈强。由于每个分子中的电子和原子核皆处在不断的运动中,因此,经常会发生电子云和原子核之间的瞬时相对位移,结果产生了瞬时偶极。两个瞬时偶极必然处于异极相邻的状态,而相互吸引,称为色散力。
本质
色散力可以采用超分子方法和对称匹配微扰理论(SAPT)进行研究。通过对几种非传统氢键化合物和传统卤键化合物的研究,是诱导力和色散力对总的吸引相互作用能占主导地位。通过卤素与呋喃分子的研究,在Π-氢键型的复合物的分子间相互作用中,色散力和静电力对复合物的形成过程中起到了关键作用。由此推论,并通过与噻吩分子之间的研究,当元素为氟和氧时,静电力为主要的能量,当元素为氮、氯、硫、磷时,色散力为主要能量,原因是与噻吩分子之间形成了Π-氢键型,并由一定的空间构型。
测量方法
1930年,菲列兹·伦敦所提出的公式中表示了色散力的可加性,即三个分子的色散能可以从三个成对相互作用的和中计算出来。但是,这个结论并没有被更复杂的范德华力相互作用所证实,它们通常不被认为是成对加性的,两个分子之间的力受到周围其他分子的影响,因此不能简单地将所有成对相互作用相加来确定一个分子与其他分子的总相互作用能。虽然与非可加性的偏差可能很小,但对于大分子,必须考虑到这一点。
分子间相互作用中的色散力的测量可以采用比较传统的两种方法:(1)对称匹配微扰理论(SAPT)和超分子方法在SAPT中,必须预先确定分子片段,并通过对分子复合物的扰动计算相互作用能。其优点是能量来自于物理上有意义的成分,这也便于定性解释。然而,SAPT只适用于碎片之间的相互作用很小,并且可以扰动地处理的样品中,这就排除了对共价连接片段的应用在超分子方法中,能量被简单地计算为复合物能量与单体能量之间的差,但是只适用于非共价配合物。(2)能量分解分析(EDA)也可用于量化色散力对相互作用力的测量,将等距反应格式应用于参考状态难以定义的系统也具有指导意义。
双波函数(WFT)方法是测量色散力的简单方法,但是,这种方法是以牺牲部分准确性为代价的。微扰理论对饱和体系和氢键体系能计算精确的能量,但是,这种方法在不饱和体系的应用中失败了。最近发展的一种常用的可视化色散力的工具是非共价相互作用(NCI)分析该方法与巴德的分子中的原子量子理论(QTAIM)密切相关,该理论已被特定于非共价相互作用的应用。关于确定色散相互作用的相对权重的波函数的分析也可以用价键理论来完成,价键理论揭示了相互作用的分子全部或部分之间的电荷转移相互作用,符合菲列兹·伦敦最初公式中振荡偶极子的计算,这种方法适用于小分子,要想计算大分子的色散力,需要将主要相互作用的C-H⋅⋅⋅H-C键与分子中电子重新耦合计算。
应用
色散力的累计
色散力是范德华相互力作用中最弱的一部分。它们产生于相邻原子上瞬时诱导偶极子之间的吸引力,是一种弱相互作用,导致它们在分子稳定性和反应性的重要作用被忽视。色散力在分子与环境的相互作用中是最常见的也是无处不在的作用力,不管是作为分子内还是分子间作用力。实际上,在生物大分子或化学超分子中,由于色散力会随着体积而累加,因此在这些物质中色散力很强,在有的分子中甚至可以和氢键强度相当。
色散力在离子液体研究中的应用
在工业中离子液体的分析
离子液体在工业上是一种常见的溶剂,广泛应用于催化重组等工业反应,探究离子液体中的非共价相互作用对于精细化控制化学反应的进程是十分重要的。离子液体中,作用更多的是库仑力,但是在非质子离子液体中的局部氢键和定向氢键可以导致更多的流动性而不是更粘稠的液体,对于蒸发焓等热力学性质,可以观察到它们几乎随咪唑阳离子烷基链长度的增加而线性增加。与正构烷烃和正醇的比较表明,每个亚甲基的分子间相互作用强度的线性增加仅仅是色散力影响得到的结果。但是氢键相对色散力比较容易分析,于是在一组离子液体中,包括三烷基铵阳离子和甲基磺酸盐和三氟酸盐阴离子,检测其中中阳离子和阴离子之间由氢键和色散力主导的相互作用的转移,通过远红外光谱中检测两种离子的特征振动峰的分析,可以直接获得体系中氢键和色散力的相对强度。经过计算在铵阳离子的烷基链上,每增加一个亚甲基,色散力增加约为2.3 kJ mol。
卤代咪唑离子液体的分析
卤代咪唑离子液体是比较常用的离子液体,以一系列基于咪唑分子的复合物为研究对象,对三种不同类型的卤键作用(离子对型卤键、电荷辅助卤键和中性卤键)进行了计算,通过能量分解分析(EDA)表明,离子对型卤键的吸引作用由静电力主导,而色散力的贡献则很小。与之不同,在中性卤键中,静电力与色散力的贡献几乎相当。值得注意的是,诱导作用(对应于电荷转移及轨道相互作用)在离子对型卤键和电荷辅助的卤键中也起到重要作用。根据计算结果,氯代咪唑阳离子与阴离子之间的作用力较弱,因此可以把氯代咪唑阳离子作为模板结构来设计新型的室温卤代咪唑类离子液体。
咪唑离子液体在太赫兹屏蔽方面的应用
咪唑及其异构体吡唑在太赫兹波段具有的独特光谱特征,使该类物质具有一定的太赫兹屏蔽作用。为了进一步解释太赫兹光谱屏蔽的产生的机理,通过密度泛函理论对咪唑和吡唑进行了分子结构建模与优化,采用势能分布分析确定了不同特征吸收峰的振动特征,并采用相互作用区域指示函数图形化分析、基于分子力场的能量分解分析和电子密度拓扑分析相结合的方法定性和定量地分析它们的分子间弱相互作用,获得咪唑和吡唑的分子结构和分子间弱相互作用等微观结构与太赫兹特征振动模式的关系,从振动特征差异和分子间弱相互作用差异的角度,解释了太赫兹光谱的吸收特性和色散力特性,进而揭示了太赫兹特征吸收峰的产生机理。
色散力对烷烃计算的应用
正烷烃分子间的作用包括各向同性的色散力和各向异性的色散力,光散射实验,量热和表面张力的测定都证实了正烷烃中存在着各向异性的色散力,它是由于相邻分子平行取向而造成的这种相关分子的定向作用在长链烃中表现强烈, 在环烷烃, 高支链烷烃以及六个碳以下的低级烷烃中可以忽略不计。但是在各向同性与各向异性的色散力作用下,对于正烷烃的内聚能和定向作用能的计算是重要的。直链烷烃分子是非极性分子,分子中碳原子越多,色散力越大,也越不容易脱离液相。因此直链烷烃的沸点随着式量的增大而有规律的升高,色散力只有在近距离内作用才明显。它随着距离的增大很快减小,有直链的分子由于支链的阻碍,不能紧密地靠在一起。因此,带有支链的烷烃分子的色散力比直链烷烃小,沸点也相应的低,且支链越多,相应的作用力也越小,沸点也越小。
色散力对催化反应的影响
色散力在催化反应中是一个决定性的因素,对其他反应性更强的碳氢化合物和一些取代体系的热力学稳定性也是一个决定性的因素。溶液中分子平衡的相对稳定性是由分子内色散力、溶质/溶剂相互作用和熵效应的微妙平衡所决定的,它们部分地相互抵消。但值得注意的是,分子内色散力效应是比较明显的,以至于它们总是使空间上更拥挤的异构体更稳定,使催化反应更容易进行。
色散力可以被应用于催化反应原理的解释中,采用密度泛函理论方法对铑(II)催化的重氮氧化吲哚与苯乙烯间的环丙烷化,反应的机理和立体选择性进行了细致的理论研究,考察了非催化反应、Rh2(OAc)4催化的外消旋反应以及手性Rh2(S-PTTL)4催化的不对称反应三类体系。结果表明非催化环丙烷化反应为能垒很高且选择性很差的单步协同反应,而 Rh2(OAc)4 和Rh2(S-PTTL)4催化的环丙烷化反应为分步过程,主要包括卡宾生成和环丙烷化两步,且环丙烷化步为协同不同步的单步过程。结果表明N脱去为催化环的决速步而环丙烷化为立体选择性决定步。计算表明,苯乙烯可沿两种路径进攻卡宾络合物,且前者比后者更有利。这项研究建立了合理的立体化学模型并成功预测了反应的立体选择性。计算探明了 Rh2(S-PTTL)4催化剂优秀的催化活性和立体控制能力的本质,结果显示非共价相互作用(NCI)在提高反应活性和控制立体选择性方面起到重要作用。同时,这种方法学研究也表明考虑色散作用对于催化反应机理和立体选择性十分重要。
色散力在絮凝中的应用
絮凝技术在中药水提液除杂纯化中具有重要的应用价值,但由于中药液种类繁多、成分复杂,药液中杂质与絮凝剂的相互作用及絮凝机理尚不明确,导致杂质絮凝效果和絮凝效率难以大幅提高。对中药水的絮凝处理实验进行分析,通过量子化模型进行模拟研究,从分子层面揭示杂质絮凝发生的推动力、杂质和絮凝剂相互作用的活性点位以及不同官能团对絮凝相互作用的贡献,得出结论,通过热力学参数和絮体复合物构型分析,发现范德华力(含诱导力、色散力)和氢键作用是絮体形成的主要推动力,杂质与絮凝剂之间的相互作用可解构为静电作用、交换互斥作用、诱导作用和色散作用,其中,静电作用是最主要的相互作用,在总吸引能中占比依次为53.21%、49.58%、52.77%、54.48%和57.50%,诱导和色散作用也是不可忽略的重要组成部分。
色散力在水处理方面的应用
色散力相关的文章

郭靖,是金庸武侠小说《射雕英雄传》男主角和《神雕侠侣》中的重要角色,《倚天屠龙记》中也曾引述其相关事迹,他是贯通"射雕三部曲"的关键人物之一。融合“降龙十八掌”、“九阴真经”和“左右互搏”三大盖世武功为一体,遂翩然翱翔,武林尊为“天下第一侠士”,黑白二道俯首称臣,号令武林群雄,率领群雄守护着南宋襄阳
郭芙是金庸小说《射雕英雄传》和《神雕侠侣》中的角色。她是郭靖和黄蓉的长女,也是东邪黄药师的外孙女,桃花岛的第三代继承人。她和杨过从小就相识,并一起成长。郭芙受到外公黄药师的珍视,得到父亲郭靖的信任,与黄蓉母女关系深厚,被称为"芙蓉"。她出生在桃花岛,儿童时期随父母到嘉兴,曾救助过武氏兄弟和杨过等人。
陆无双,金庸小说《神雕侠侣》中的人物。原是江南陆家庄的千金小姐,陆家庄二庄主陆立鼎的女儿,幼时遭“赤练仙子”李莫愁灭门,并被其掳去,后成为李莫愁的徒弟,长大后偶遇杨过,并对杨过暗暗倾心。
开罗(英文:Cairo;阿拉伯文:قـــاهــرة),是埃及的首都,同时也是埃及、非洲及阿拉伯世界最大的城市。城市横跨尼罗河,为整个中东地区的政治、经济、文化和交通中心,位于埃及的东北部。开罗占地3085km²,人口数量2280万(2017年),拥有著名景点金字塔群,埃及博物馆,解放广场。
查士丁尼一世(又译优士丁尼一世,拉丁文:Iustinianus I;希腊文:Ιουστινιανός Α´;约482年-565年11月14日),东罗马帝国皇帝(527-565),史称查士丁尼大帝(英文:Justinian the Great)。

尚可名片
这家伙太懒了,什么都没写!
作者

